Proteins are the molecules of life, essential for a wide range of functions from protecting the body from viruses to building the structures of all living things. Protein engineering has been used to repurpose these machines, and proteins can be optimized for new, valuable functions for technological, scientific, and medical applications. New approaches to protein engineering leverage machine learning to find the best proteins for a given application with limited experimental characterization. A machine learning model can be used to predict how well a protein performs a given task, in other words, its “fitness”. The way in which we computationally represent proteins is crucial in determining how these machine learning models predict protein properties. While most approaches use a protein’s sequence to represent proteins, we hypothesize that protein structure provides richer information for the model to learn these properties. We represent each protein of interest as a graph, or a network of amino-acid connections in the protein, and implement a graph machine learning model to predict a protein’s fitness. We show that this structure-based model has superior performance to sequence-only approaches for fitness prediction. We further extend this model to automatically find the best protein for a given task by optimizing a protein’s graph representation. We demonstrate that this novel approach is the most successful in finding the best protein from a small number of proposed proteins.
Author: Marianne Arriola
University of California, Santa Barbara
Mentors: Frances Arnold and Kadina Johnston
California Institute of Technology
Editor: Wesley Huang
Using Machine Learning to Explore the Non-natural World
Protein engineering has been used to uncover and optimize protein sequences that are not found in the natural world but have valuable functions for technological, scientific, and medical applications [1]. Protein engineers hope to find proteins with maximal “fitness,” a measure of how well a protein performs a desired function – for example, proteins that are strongest at enacting responses for disease treatment or proteins that are the most stable in different environments. Machine learning has accelerated the lab-based search for optimal proteins by computationally predicting protein fitness [1].
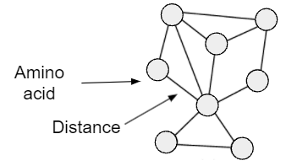
To predict fitness, one represents the protein in a data structure understood by a model so that it extracts as much useful information about the protein as possible. Informative protein representations are essential; they determine how the model learns from a protein’s characteristics to predict its fitness. The most common approaches represent proteins as sequences of amino acids, the basic building blocks of proteins [1]. However, protein structure is crucial for function and therefore may better represent proteins by encoding how a protein twists, folds, and bends to determine its function. Recent work demonstrates that graphs, a data structure used to describe relationships between entities, efficiently represent protein structure [3]. A protein can be instantiated in a graph through nodes (amino acids) and connecting edges (distances between amino acids) (Figure 1). Previous work has found that these representations provide higher accuracy in predicting structure-function relationships than sequence-based representations [3]. Though graph representations have been used to classify protein function, to the best of our knowledge they have not yet been used to predict protein fitness. Therefore, we propose graph-based approaches to represent and evaluate proteins.
Research objectives
We test whether structure provides more information than sequence for fitness prediction
We hypothesize that a structure-based approach will result in higher predictive accuracy than sequence-based approaches because structure directly impacts protein function and therefore may provide more useful information to predict fitness.We implemented a graph convolutional network (GCN), a machine learning model that learns on graph-structured data, to predict protein fitness using structural information which would enable us to identify high-functioning proteins. We then measured the advantage that a structure-based prediction model has over a sequence-based one by comparing their predictive performances. Moreover, this approach can validate or potentially improve upon computational fitness prediction in the Arnold Lab for optimal protein identification.
We explore whether fitness prediction models can predict activity on different substrates
Protein fitness is dependent on the identity of the substrate, or protein-activating molecule, that the protein performs activity on. We propose that activity on one substrate cannot predict the activity on other substrates because a machine-learning model is tuned and trained based on the patterns in the data it is given (in this case, activity on a particular substrate). The model can have less predictive accuracy when given data with different trends (activity on alternate substrates).
Substrate structure is also crucial to protein activity: protein activity can vary widely in response to subtle changes in substrate structure. When nature has evolved a protein for a specific substrate, that activity is referred to as natural activity. Activity on substrates with fundamentally different structures is termed non-natural activity, and, importantly, it is an activity that nature has not evolved for and must be engineered. We explored model performance of predicting non-natural protein activity compared to natural activity. A model that can accurately predict non-natural activity is valuable because protein engineers typically pursue activities which nature has not yet optimized.
We attempt to automate optimal protein identification
We define a novel method to efficiently search through spaces of large numbers of variants using a protein’s graph representation. We believe that this approach could enable automated discovery of optimal proteins for a given function without having to explore every possible variant, which is computationally expensive. The model should deduce which structural patterns result in higher fitness to find the optimal protein.
Methods
How do we test our models?
We tested our approaches to find the optimal protein amongst all ~6000 single variants ⎼ variations of a protein with a mutation at a single amino acid position in the sequence ⎼ of amiE, a gene that encodes an enzyme from P. aeruginosa [4]. Wrenbeck et. al quantified the fitnesses of these variants (cell growth, in this case) when performing activity on three substrates: acetamide (ACT), propionamide (PR), and isobutyramide (IB). Activity on ACT and PR is considered more natural, as they have structures for which amiE has been naturally evolved, than comparatively unnatural activity on IB, which has a fundamentally different structure [4].
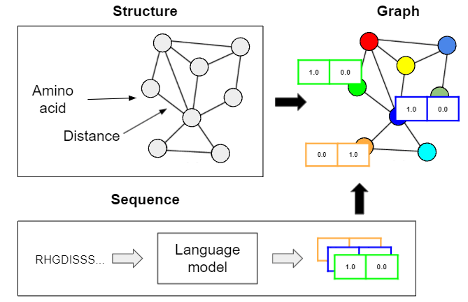
How can we encode protein structure into a graph?
We used Triad, a physics-based protein modeling software, to predict protein structure from sequence. We then used the predicted structure to generate its graph representation that describes amino-acid contacts in the protein. We applied a language model, a model architecture that typically learns from text (in this case, protein sequence) [3], to extract amino acid-level features used to describe the nodes of the graph representation. Figure 2 demonstrates the construction of the protein’s graph representation, where structure and sequence information are used in tandem.
Can structure-based representations improve fitness predictions?
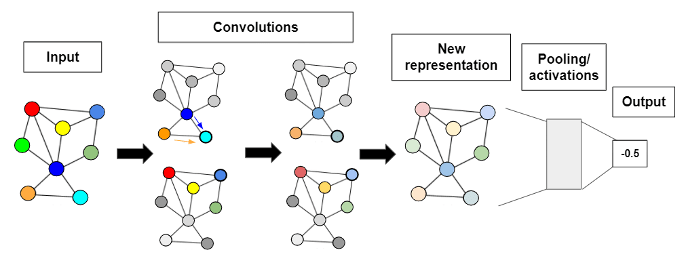
We trained a GCN to learn and subsequently predict protein fitness from its graph representation, as illustrated in Figure 3. This model takes in a graph as an input that represents some protein then passes it to a series of convolutions. These convolutions uncover potentially valuable hidden information about the protein by increasing the complexity of the graph representation via message passing, a framework in which node information is passed to connecting nodes at each step. In this context, sequence information about each amino acid is used to update the information describing other amino acids. The GCN produces a new, higher-level graph representation where each node not only contains information about itself, but also about its neighbors. This is followed by a pooling layer and an activation function that reduce the dimensionality of the graph to a single fitness score. The model ‘learns’ to output an accurate fitness score by learning which node connections to assign more priority to in the convolutions that result in more accurate prediction.
How can one efficiently navigate a protein fitness landscape using structure?
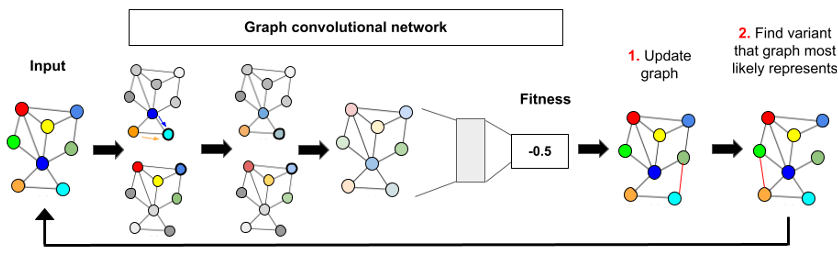
We defined a method to efficiently find the optimal protein rather than predicting the fitnesses of all candidates, which is computationally expensive. To do so, we only predict the fitness of a smaller subset of the variants: we optimize the graph structure to retrieve the one that has the highest predicted fitness. We modified a protein’s graph representation to find a new one in the “landscape” of all potential protein variants that is likely to increase predicted fitness until the protein with the best fitness is found (Figure 4) [5].
Results
Structure-informed representations improve fitness predictions
Our hypothesis was confirmed by comparing the performance of structure-based and sequence-based fitness prediction models. Performance here is measured by the correlation between true and predicted fitnesses: high correlation (above 0.5) means that the model can accurately identify high-functioning and low-functioning variants. Representations that utilize structure yield higher performance than those that solely rely on sequence (Figure 5). This demonstrates that structure-informed protein fitness prediction models are more accurate than the baseline sequence-based approach. This confirms that protein structure is more informative than sequence.

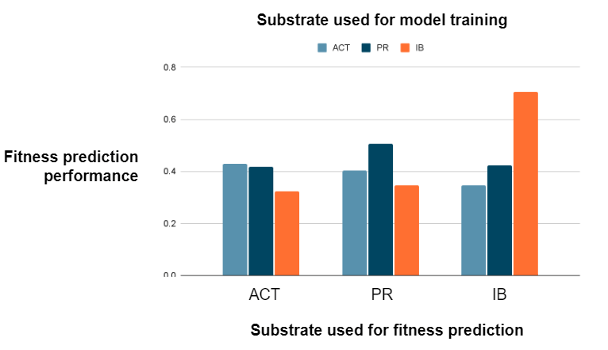
Substrate identity results in independent fitness predictions
We show that enzyme activity is substrate-specific as the fitness prediction model performs poorly at predicting the activity on different substrates other than the one that is used for model training. As Figure 6 shows, the model that performs best at predicting the activity on a given substrate is the model that is trained on activity on that substrate. As fitness predictions on one substrate cannot be transferred to another, enzyme activity must be substrate-specific.
Furthermore, for the model that performs best at predicting activity on a given substrate, our approach is best at predicting non-natural activity on IB rather than natural activity on ACT and PR. Thus, our results imply that our model can best predict non-natural activity. We suggest that this is due to the fact that IB has a wider range of fitness values across variants compared to activity on ACT and PR; a wider range of fitness values means that the model can learn more structure-fitness patterns whereas a small range limits what the model can learn.
Graph optimization can identify optimal proteins in small fitness landscapes
We designed a method for finding proteins of optimal fitness by optimizing and repeatedly updating the graph representation of the protein. We found that in a small landscape of 30 variants, the model could successfully identify the variant with the optimal fitness. Figure 7 shows the updated representation’s similarity to the optimal variant throughout training: ideally, the model would produce a variant that is identical to the optimal variant, with similarity increasing as training round increases. As shown in Figure 7 (left) the model navigates to the optimal protein at around the 35th training round, where the similarity oscillates between the variant the model deems to have ideal fitness and another variant in the landscape. This pattern signifies that the model has already found a protein that it deems to be optimal and can no longer navigate the landscape to find new variants.
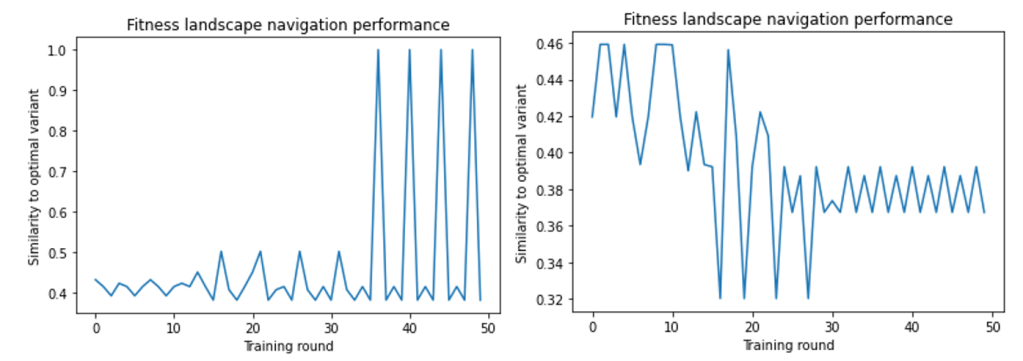
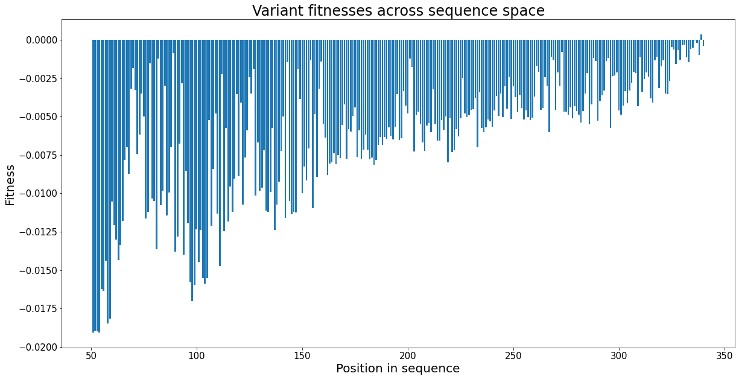
However, in a larger landscape of 600 variants, the model finds a protein with higher fitness, but not the variant with 100% similarity to the optimal, as shown in Figure 7 (right). This is likely because the model finds a protein with maximum fitness relative to proteins nearby in sequence space rather than relative to the entire landscape. Figure 8 shows the distribution of fitnesses across sequence space, where there are multiple local maxima in fitness scattered throughout the landscape that the model can incorrectly identify as the optimal protein.
Discussion
Structure-based fitness prediction improves over a sequence-based approach, making it a promising fitness prediction tool that is more accurate for finding optimal proteins for a given function. However, there are limitations: it would be computationally expensive to compute the structure for every variant rather than simply using sequence for fitness prediction depending on the number of variants. The user must assess the number of variants that will be used as it may be of less interest to use a graph-based approach for a larger number of variants.
We found that the structure-based model performs considerably better at predicting non-natural activity than natural activity. This implies that the model can be utilized in predicting the fitnesses of potentially valuable non-natural proteins that can be created via protein engineering.
This work has only been tested on a single-mutant fitness landscape, where there are only small changes in structure. However, it may be of interest to search for the best protein through variants with multiple mutations, where there are larger changes in structure. If there are larger changes in structure, the model may have more success at differentiating between structures in producing fitness predictions. Unfortunately, landscapes with multiple simultaneous mutations are largely populated by variants that exhibit little to no activity (extremely low fitness), making navigation a more difficult task.
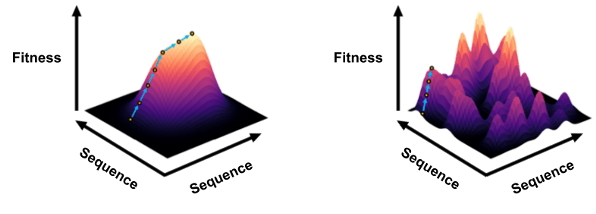
Fitness landscape navigation is of interest as it would allow one to automatically find optimal proteins for a given function without lab experimentation and without calculating every candidate’s fitness. This is a novel approach in machine learning for fitness prediction because most related approaches only focus on predicting a given protein’s fitness. However, navigating diverse landscapes with different topologies may pose challenges. Our landscape navigation method aims to incrementally find proteins with higher fitness until it finds the optimal protein (in other words, the highest peak in the landscape) as shown in Figure 9. The navigation model proposed here incrementally finds better proteins until it has found a peak which it assumes is the optimal protein, but there is no way of guaranteeing that it is the highest peak (the true optimal protein) without exploring every point in the landscape. This makes the model successful in finding the optimal protein in a landscape with only one peak (Figure 9, left). However, if there are multiple peaks, the model may find a peak that is not the highest and incorrectly assume that it is the optimal protein (Figure 9, right).
Our fitness landscape navigation model can successfully find the optimal protein in a small landscape, but it was unsuccessful in a larger landscape likely because it is more complex and has multiple peaks. Future work to make this navigation robust to diverse landscapes will give researchers freedom to find any optimal protein, no matter how smooth or rugged the landscape is.
Conclusion
In summary, we first found that protein structure is more informative than its sequence for protein fitness prediction. We also found that non-natural activity is more predictable than natural activity. Third, we showed that a fitness prediction model trained on a specific substrate does best at predicting activity on that substrate. Finally, the fitness landscape navigation model proposed here to automatically find the best protein for a given task succeeds in smaller landscapes but struggles in larger, complex landscapes.
It is worth exploring this approach further to enable successful navigation of larger, diverse fitness landscapes: for example, landscapes of variants that have multiple mutations. Navigating through variants with multiple simultaneous mutations is likely to result in barren landscapes with few peaks; it may be of interest to define a method to navigate through such landscapes. A potential approach would begin several navigation searches at different points in sequence space, with the hope that the model will find different peaks.
This work is a promising step in informed protein engineering, which will enable better protein identification needed for impactful technological, scientific, and medical applications.
Further Reading
Exploring protein fitness landscapes by directed evolution
Machine-learning guided directed evolution for protein engineering
Structure-based protein function prediction using graph convolutional networks
References
- Yang, K. K., Wu, Z. & Arnold, F. H. (2019) Machine-learning-guided directed evolution for protein engineering. Nat Methods 16, 687–694. DOI: 10.1038/s41592-019-0496-6
- Romero, P., Arnold, F. (2009) Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol 10, 866–876 DOI: 10.1038/nrm2805
- Gligorijević, V., Renfrew, P.D., Kosciolek, T. et al. (2021) Structure-based protein function prediction using graph convolutional networks. Nat Commun 12, 3168. DOI: 10.1038/s41467-021-23303-9
- Wrenbeck, E., Azouz, L. & Whitehead, T. (2017) Single-mutation fitness landscapes for an enzyme on multiple substrates reveal specificity is globally encoded. Nat Comm 8, 15695. DOI: 10.1038/ncomms15695
- Li, J., Tianyun Z., Hao T., Shengmin J., Makan F., and Reza Z. (2020) SGCN: A Graph Sparsifier Based on Graph Convolutional Networks. Advances in Knowledge Discovery and Data Mining 12084: 275.
Acknowledgments
I would like to thank my co-mentor, Kadina, for being supportive of my ideas, patient with my work, and for giving me valuable insights on my project; I would also like to thank my mentor, Dr. Frances Arnold, for giving me the opportunity to work outside of my discipline in chemical engineering research, and Dr. Sabine Brinkmann-Chen for taking the time to give helpful feedback on my reports and presentations. I thank my principal investigator from the DYNAMO Lab at UCSB, Dr. Ambuj Singh, for inspiring me to pursue graph machine learning. Finally, I send my thanks to the Resnick Sustainability Institute (RSI) and Student-Faculty Programs (SFP) at Caltech for providing me with financial support on my project.
