Author: Stella Baldwin
Mentors: Dr. Victoria Orphan, Dr. Daniel Utter
Editor: Suchitra Dara
Abstract
The factors controlling microbial diversification remain relatively unknown, particularly in deep sea environments like methane seeps. Here, sediment-dwelling anaerobic methanotrophic archaea (ANME) pair symbiotically with sulfate-reducing bacteria (SRB) to fuel the anaerobic oxidation of methane (AOM) and other biologically productive processes. ANME-2b are a poorly understood clade abundant in cold methane seep sediments like those off the coasts of Southern California and Central America. Previous work identified three subgroups within ANME-2b-gI, gII, and gIII- and hypothesized that only gII and gIII contain nifH, a gene that encodes an essential nitrogen fixation protein. However, experimental validation of this hypothesis has proven difficult due to the lack of strain-resolved microscopy approaches. In our case, strain-resolved microscopy involves visualizing microbial genetic variations with fluorescent probes. Here, we investigated fluorescence in-situ hybridization (FISH) techniques to help visualize ANME-2b lineages. Genome-directed FISH offers extreme specificity but low signal, so we chose a signal amplification method called hybridization chain reaction FISH (HCR-FISH). We improved HCR-FISH probe contrast by testing complex samples under different temperatures. HCR-FISH probe validation requires known positive/negative targets, so we used a natural deletion in the model archaeon Methanosarcina acetivorans as a control system. However, we found that experimental parameters like hybridization temperature were not directly transferable between ANME-2b and M. acetivorans. We concluded that HCR-FISH optimization is best conducted on the desired organism/system. Once refined, genomic HCR-FISH could illuminate the intraspecific ecology and evolution of many microbial systems.
Introduction
Methane seeps are natural seafloor gas leaks that fuel high primary productivity (high rates of biomass production), deep ocean nutrient cycling, and biodiversity hotspots 1. The National Oceanic and Atmospheric Administration (NOAA) identified methane seeps as essential fish habitats, providing economic benefits on the United States Pacific Coast where they are located. Further, seep methane cycling contributes substantially to Earth’s carbon budget. The anaerobic oxidation of methane (AOM) in methane seeps consumes over 7-25% of global methane production 3. This AOM is performed by anaerobic methanotrophic archaea (ANME) paired with specific symbiotic sulfate-reducing bacteria (SRB) 3. These organisms typically perform AOM through the following reaction: CH4 + SO42− → HCO3– + HS− + H2O 3. ANME/SRB pairs make AOM more efficient. These pairs halve the AOM chemical reaction: ANME oxidize methane and transfer the electrons to SRB; SRB then reduce sulfate with those electrons. As chemoautotrophs, methane-oxidizing microorganisms supply energy to methane seep microbiomes 1. Despite archaeal ubiquity, identifying the significant environmental factors that control microbial evolution remains challenging. Yet, identification is a prerequisite for understanding the evolution and resilience of these important microbial communities. Here, we study evolution in methane seep communities to better understand critical microbial organization mechanics (i.e., methane and nitrogen cycling).
Despite evolving in such a thermodynamically constrained niche, ANME encompass tremendous diversity spanning multiple orders4 . Microbiologists initially hypothesized that ANME-SRB symbioses based on AOM could not support microbial life due to a low thermodynamic yield (-16 kJ/mol) 3. But sediments from Eel River Basin, the Black Sea, and Hydrate Ridge revealed abundant ANME and sulfur-reducing bacteria (SRB) symbioses 3. These ANME with varied syntrophic associations and biogeochemical habitats fall into 3 main groups: ANME-1, ANME-2, and ANME-3. 16S rRNA genes reveal that AMNE clades fail to be monophyletic (classified in the same taxon) since sequence similarities vary between 75-92% 3. However, little is known about ANME growth with low energy yields and AOM constraints. For instance, what biotic or abiotic factors drive the evolution of AOM consortia? At what spatial and phylogenetic scales are different AOM consortia functionally interchangeable vs. distinct? How will climate change affect AOM processes? Researching the diversity of methanotrophic and methanogenic archaea via 16S rRNA and metabolic genes (e.g., mcrA, the gene that encodes a methyl coenzyme M reductase, a key step in methane oxidation) could answer these questions.
The Orphan Lab currently researches active methane seep sites off the Santa Monica Basin, which are produced by tectonic activity between the North American and Pacific Plates. Here, we focus on ANME-2, which dominate cold seep near-surface sediments off the coast of Southern California and couple with the SRB Desulfococcus 3. ANME-2 contain the most diverse ANME clades, subdividing into 4 groups (2a-2d) that roughly correspond to different genus or family designations 4. ANME-2a-c are united by their seemingly obligate association with SRB partners reflected in their co-diversification 5. Amino acid identities of one group, ANME-2b, vary significantly, suggesting several distinct species 4. Within ANME-2b, the Orphan Lab identifies three ANME-2b groups: gI, gII, and gIII. We hypothesize that only gII and gIII contain nifH, a gene encoding an essential protein for nitrogen fixation.
The Orphan Lab next aims to increase understanding of ANME-2b ecological and evolutionary diversity using an HCR-FISH microscopy approach. Traditional ribosome-directed fluorescence in-situ hybridization (FISH) historically fails to provide enough resolution between closely related strains, like those within ANME-2b, due to the slow-evolving nature of the 16S rRNA gene. Therefore, hybridization chain reaction FISH (HCR-FISH) is preferred, as it can be employed as a genome-directed method to sample faster-evolving genes. Because genome-directed FISH lacks the inherent amplification that ribosome-directed FISH approaches enjoy, HCR-FISH incorporates an amplification step after the initial probe hybridization. HCR-FISH is a two-step process (Figure 1). Step one involves adding an unlabeled probe targeting a specific nucleotide sequence. Step two involves hybridizing a fluorescent secondary probe to the first. The secondary probe forms a hairpin structure that unfolds once hybridized to a primary probe. The secondary probe presents binding sites for hairpin hybridizations, allowing for the chain reaction to amplify the single primary probe signal. A successful HCR-FISH probe has good contrast and specificity under the microscope. If successful, HCR-FISH tools will allow spatial mapping of ANME-2b partnerships, roles, and phenotypes.
Our project helps develop HCR-FISH microscopy techniques for ANME-2b lineage identification. We test three groups: ANME, Methanosarcina acetivorans, and Santa Monica methane seep microbial mats. We begin with ANME HCR-FISH experiments to improve ANME-2b microbial lineage visualization. We optimize contrast by testing HCR-FISH probes on complex samples in different temperature conditions. However, specificity requires a defined community with both known positive and negative targets. Little is known about ANME, so positive and negative targets were hard to identify. Thus, we also employ HCR-FISH experiments in Methanosarcina acetivorans, a model methanogenic archaeon previously analyzed by the Orphan Lab. We leverage a spontaneous M. acetivorans mutation (21 kbp deletion, resulting in a strain nicknamed “Franken”sarcina) to test known genomic regions. Unlike M. acetivorans, “Franken”sarcina lacks many genes, including those required for growth on acetate, due to the deletion. However, both contain the mcrA gene that aids in methanogenesis catalyzation. By testing probes on genomic differences between M. acetivorans and “Franken”sarcina, we can modify HCR-FISH probes to better identify positive and negative targets. Additionally, we use FISH on Santa Monica mat samples from the July 2023 AT50-12 cruise off the coast of Southern California. Once HCR-FISH is improved, this project will help identify AMNE-2b evolution and specify other microbial systems with closely related yet ecologically distinct lineages.

Results
ANME HCR-FISH is optimal at 46 °C. The ANME experiments were designed to test the hypothesis that all ANME-2b gII, but not all gIII, cells contained nifH genes. Thus, our initial probeset included three HCR-FISH probe mixes which were each computationally predicted to be specific to gII and gIII and to nifH. The results indicated gII and gIII probes bound to a subset of the total aggregates seen by DAPI and fluoresced greater than background, but nifH signal was absent (Figure 2). Negative no-probe controls were also performed to account for autofluorescence. Based on these results, we wanted to confirm 1) whether the gII or gIII probes were indeed specific to ANME cells and 2) whether the signal/background contrast could be increased. We replaced the gIII probe mix with a general Archaea probe (Ar915) and found that gII probes were specific to Archaea cells rather than SRB or other microbes based on overlap between the Ar915 and gII probes. Melting double-stranded genomic DNA could allow primary probes better access to binding sites, so we also varied the temperature for the initial hybridization step to 46 °C and 80 °C. At 46 °C, gII showed more ANME signal/noise contrast than 37 °C (Figure 3). 80 °C failed, showing similar results to the control. However, nifH was not detected, suggesting that the signal is still too dim or the nifH probe mix needs to be refined. Our results indicate that ANME HCR-FISH probes provide better signal to background contrast under 46 °C incubations, but beyond a certain point (i.e., 80 °C) microbes may degrade.


M. acetivorans and “Franken”sarcina HCR-FISH uncover unexpected genomic rearrangement. To continue optimizing genome-directed HCR-FISH, we turned to a model system of closely related M. acetivorans strains in pure culture. To transfer HCR-FISH techniques to M. acetivorans, we designed genome-directed probe sets analogous to the ANME-2b groups but designed to target the differences between M. acetivorans C2A and “Franken”sarcina. Thus, we designed three HCR probe sets that targeted housekeeping genes common to both we called the “genome” set, a set targeting mcrA called “mcr”, and a set called “C2A” targeting to the C2A genes lost by “Franken”sarcina.
With these probes, we carried out four initial experiments. The first tested M. acetivorans C2A and “Franken”sarcina at 37 °C incubations without primary probes but including secondary probes. This established a no-probe control baseline for future experiments and revealed whether the HCR-FISH secondary probes were generally “sticky” to microbes or debris. The second evaluated M. acetivorans C2A and “Franken”sarcina at 37 °C with all HCR-FISH probes to determine HCR-FISH probe effectiveness. The third redid the second test but with primary probe hybridization at 46 °C based on experiments in ANME. The fourth tested M. acetivorans and “Franken”sarcina at 37 °C with only half of the B1 primary probes but all secondary probes. This assessed the relationship between HCR-FISH probe binding sites and signal intensity, which is useful for adapting this approach to other systems.
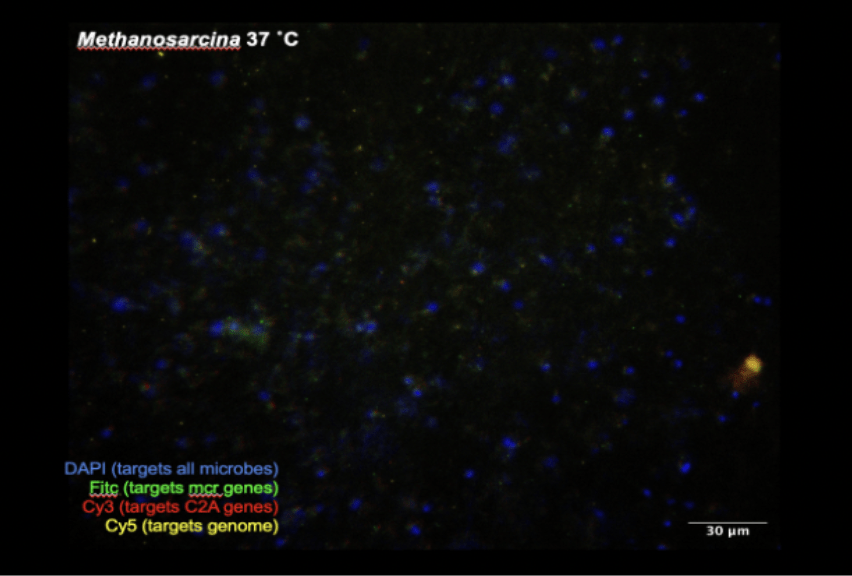
Unfortunately, all M. acetivorans and “Franken”sarcina experiments proved inconclusive. M. acetivorans and “Franken”sarcina experiments (Figure 4, Figure 6) displayed equal mcr, C2A, and genome HCR-FISH intensity when compared to their respective negative controls under identical incubation conditions (Figure 5, Figure 7), meaning secondary probes attached nonspecifically to M. acetivorans and “Franken”sarcina debris. An experimental rerun with increased wash steps produced similar results. The M. acetivorans and “Franken”sarcina experiments revealed that less is known about so-called model genomes than previously thought (Figure 8). When compared to M. acetivorans, the reconstructed “Franken”sarcina contains an unknown 156 kbp deletion and a 206 kbp addition, differing from the 21 kbp deletion theory. Contamination cannot explain this difference. These difficulties associated with transferring experimental parameters from one system to another suggests that future ANME-2b HCR-FISH optimizations will not benefit from experiments in model systems. Unlike ANME-2b, M. acetivorans and “Franken”sarcina probes did not show any improvement in 46 °C HCR-FISH incubations (Figure 9). Therefore, HCR-FISH experimental parameters are not directly transferable between ANME-2b and M. acetivorans.





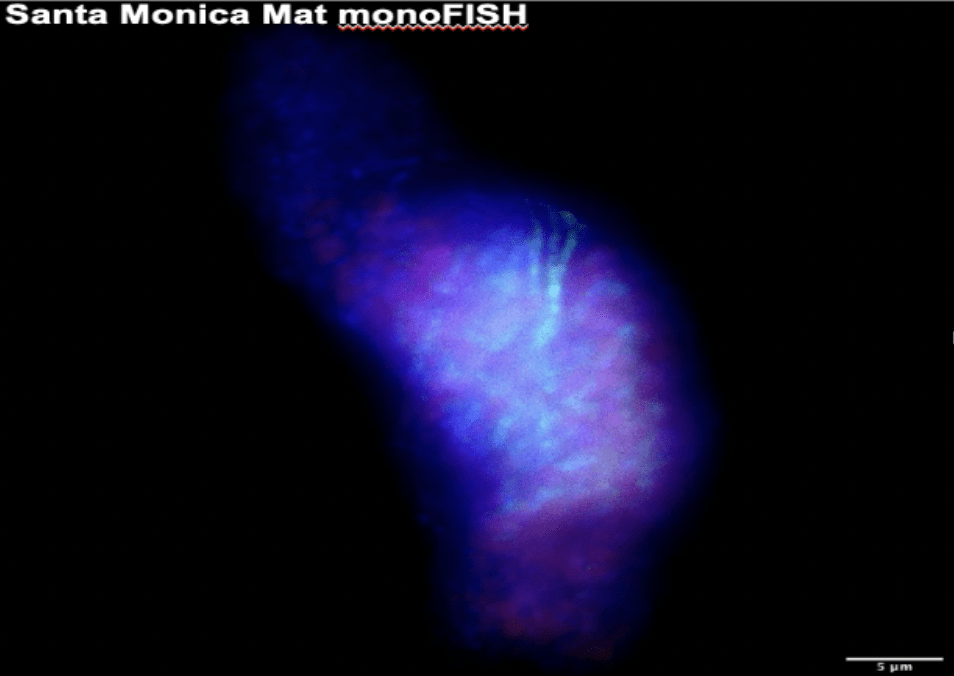
Santa Monica Basin FISH Experiment Results. Fresh samples from the AT50-12 cruise to Santa Monica Basin were surveyed with FISH experiments to ascertain the presence of ANME in seep and colonization array samples (Figure 10). This visualizes the ecological diversity thriving in the complex geochemical processes occurring there. We ran two FISH experiments with probes targeting bacteria, general archaea, and ANME-1. Each sample tested contained bacteria and general archaea hits. However, bacteria and general archaea hits sometimes overlapped, indicating either FISH probe errors or thick microbe aggregates. Bacteria hits highlighted various microbe morphologies, indicative of a variety of species. Some sediments tested also contained ANME-1 hits, but many non-ANME-1 archaea occurred in aggregate morphologies typical of ANME. Future experiments should employ probes targeting ANME-2 or ANME-3 to describe the ANME diversity more fully in these sediments.
Discussion
Our research improved microscopy approaches aimed at visualizing spatial ecology of ANME-2b phylogenetic diversity. Genomic sequencing has revealed unprecedented genomic diversity, but molecular methods to probe this predicted diversity remain underdeveloped. Thus, the big-picture objective was to visualize gene-level differences among strains via HCR-FISH. The first step was to develop HCR-FISH methods that exhibit contrast and specificity. Experiments were designed in an anticipated model system (M. acetivorans) and our original system (ANME-2b). ANME-2b experiments produced a detectable signal for two different probesets, and adjusting methodological variables led to an amplification of the signal. However, specificity between probesets was difficult to assess quantitatively in such undefined sediment enrichments. Therefore, we conducted HCR-FISH experiments in M. acetivorans to allow greater specificity controls.
These M. acetivorans experiments revealed the complications associated with transferring genome-directed FISH between systems. Probe design is based on available genome sequences, and limitations in genome quality severely impact the efficacy of probe design. We did not understand that the “Franken”sarcina genome had evolved. Therefore, our HCR-FISH probes did not stick to the intended targets. It was not our plan to reassemble the “Franken”sarcina genome, but these results emphasized the importance of understanding the target’s entire genome before using HCR-FISH probes. HCR-FISH methodology effectiveness varied between M. acetivorans and environmental systems unpredictably.
We concluded that successful HCR-FISH techniques must be modified on the organism in question. Our method, testing HCR-FISH positive and negative targets on a model M. acetivorans system, did not help improve HCR-FISH techniques in AMNE-2b. Our experiments prompt the future exploration of unknown HCR-FISH success variables. We spent the remaining weeks using FISH to assess microbial diversity in the Santa Monica Basin to compare with the prior ANME-2b experiments from off the coast of Costa Rica. Future research directions include (but are not limited to) solidifying optimal HCR-FISH incubation temperatures, further specifying ANME-2b HCR-FISH probes, and explaining new “Franken”sarcina genome variations.
Methods
ANME HCR-FISH Experiments. We began with the Orphan Lab’s “4.19.23 FISH Protocol Notes”, based off the HCR v3 protocol for E. coli 6. This protocol spanned three days: Day 1 (attach primary probe), Day 2 (attach secondary probe), and Day 3 (imaging). Sediment slurries were fixed in 2% paraformaldehyde (remainder 3X PBS) at 4 ˚C overnight.
On Day 1, the sample was vortexed, diluted into 1000 μL sterile 3X PBS, chilled, and sonicated briefly. Percoll was then added before centrifuging for 30 minutes at 4 °C to separate cells from sediment based on density. Aggregates were filtered onto a 3.0 μm polycarbonate filter and washed with cold sterile 3X PBS. The filter was cut into an irregular diamond shape to indicate which side contains the microbes. The filter was submerged in HCR-FISH hybridization buffer (Molecular Instruments, Los Angeles, CA) and incubated at 37 °C for one hour. HCR-FISH primary probes (1 μL of 2 µM stock) and/or mono-FISH probes (10 μL of 50 ng/uL stocks) probes were added to a total of 100 μL of hybridization buffer. The probe mix was then incubated overnight at 37 °C.
On Day 2, the filter was washed four times with “30% probe wash buffer V3.0” (Molecular Instruments, Los Angeles, CA) while being incubated at 37 °C. After, the filter was placed in 150 μL of Amplification Buffer (Molecular Instruments, Los Angeles, CA) at room temperature for 30 minutes. Meanwhile, light-sensitive HCR secondary probes (Molecular Instruments, Los Angeles, CA) were added to a PCR strip tube containing the amplification buffer. The PCR tube was put into a thermocycler (95 °C for 90 seconds, 22 °C for 30 minutes) to snap cool hairpins. The PCR tube contents were added to the filter, covered, and stored at room temperature overnight.
On Day 3, the filter was washed four times with SSCT buffer. Then, the filter was mounted to the slide and coated in 20 μL of DAPI-CitiFluor solution. DAPI stains DNA, and CitiFluor prevents bleaching. The filter was covered by a glass cover slip. An Olympus microscope with a 1.4 NA 100x oil immersion objective captured images. Image analysis was done with FIJI 7. After imaging, the slide was stored at room temperature in a dark slide book. We ran four similar experiments using gII B3, cy3 Ar-915 B5, and nifH B1 probes. We targeted ANME-2b gII, gIII, and nifH genes.
ANME experiments also underwent 46 °C and 80 °C overnight HCR-FISH incubations (as opposed to 37 °C on Day 1) to test ideal HCR-FISH temperatures.
M. acetivorans and “Franken”sarcina HCR-FISH Experiments. HCR-FISH in M. acetivorans and “Franken”sarcina was performed as described above but with different primary (B1 genome, B3 mcr gene, B5 C2A gene) and secondary (B1H1-647, B1H2-647, B3H1-488, B3H2-488, B5H1-560, B5H2-560) probes. Also, samples were not obtained beforehand. Therefore, the first step involved growing Methanosarcina cells anaerobically. We created a 1 mL mixture: 0.4 mL of cells, 0.5 mL of 3x PBS, and 0.1 mL of 20% PFA. We incubated the mixture or one hour at room temperature, centrifuged it three times at 12,000 rpm for 2 minutes, and washed it three times with 3x PBS.
M. acetivorans and “Franken”sarcina experiments also underwent 46 °C and 80 °C overnight HCR-FISH incubations to test ideal HCR-FISH temperatures.
Based on the poor results of the no-probe controls, an additional wash step (a new total of five washes) was performed on Day 3. No improvements occurred.
Santa Monica Mat FISH Experiments. We wanted to test microbe diversity in the AT50-12 cruise sediment samples. To do this, we ran two experiments targeting bacteria (Eub338), general archaea (Ar915), and ANME-1 (ANME-1-350) using monoFISH probes. FISH was performed according to the Orphan Lab’s “Seagrass and sediment FISH Protocol.” FISH protocol spanned two days: Day 1 (attach FISH probes) and Day 2 (imaging). AT50-12 cruise samples were stored in -80 °C laboratory refrigerators until use.
On Day 1, the sample was defrosted at room temperature, smeared onto a 75 x 25 mm microscope slide, and dried at room temperature. Once dried, each slide was dipped into three ethanol petri dish mixtures for sample dehydration. The slide was submerged in 50% ethanol for three minutes, 80% ethanol for one minute, and 90% ethanol for one minute. While each slide dried, 1 μl of each FISH probe was added to 10 μl of 40% formamide stringency hybridization buffer. The FISH mixture was added to each dehydrated sample, covering the sample entirely. 500 uL of hybridization buffer was added to tissue paper in a 50 ml screw-top plastic tube. The tightly sealed plastic tube became a moisture chamber, preventing evaporation from the slide. Each slide was added horizontally into individual 50 ml screw-top plastic tubes before incubation at 46 °C overnight.
On Day 2, the slide was washed with a 40% wash buffer for 10 minutes at 48 ˚C. Then, the slide was dipped for 30 seconds in nanopure water. 20 μL of DAPI-Citiluor solution was added to the dried slide before covering it with a glass cover slip. A Zeiss Elyra PS.1 microscope with a 1.4 NA 100x oil immersion objective captured images and Z-stacks. FIJI colorized images and merged visual data 7.
Acknowledgements
Thank you to the Orphan Lab (Dr. Victoria Orphan, Dr. Daniel Utter, Dr. Rodney Tollerson II, and Rebecca Wipfler) for mentorship, training, and resources. Thank you to the Leadership Alliance’s Summer Research Early Identification Program for guidance, funding, and preparation to pursue a future Ph.D. program. Thank you to Caltech’s Center for Environmental Microbial Interactions (CEMI) WAVE Fellowship for funding.
References
- Ruff, S. E., Biddle, J. F., Teske, A. P., Knittel, K., Boetius, A., & Ramette, A. (2015). Global dispersion and local diversification of the methane seep microbiome. Proceedings of the National Academy of Sciences, 112(13), 4015–4020. https://doi.org/10.1073/pnas.1421865112
- Paull, C. K., Hecker, B., Commeau, R., Freeman-Lynde, R. P., Neumann, C., Corso, W. P., Golubic, S., Hook, J. E., Sikes, E., & Curray, J. (1984). Biological Communities at the Florida Escarpment Resemble Hydrothermal Vent Taxa. Science, 226(4677), 965–967. https://doi.org/10.1126/science.226.4677.965
- Knittel, K., & Boetius, A. (2009). Anaerobic Oxidation of Methane: Progress with an Unknown Process. Annual Review of Microbiology, 63(1), 311–334. https://doi.org/10.1146/annurev.micro.61.080706.093130
- Chadwick, G. L., Skennerton, C. T., Laso-Pérez, R., Leu, A. O., Speth, D. R., Yu, H., Morgan-Lang, C., Hatzenpichler, R., Goudeau, D., Malmstrom, R., Brazelton, W. J., Woyke, T., Hallam, S. J., Tyson, G. W., Wegener, G., Boetius, A., & Orphan, V. J. (2022). Comparative genomics reveals electron transfer and syntrophic mechanisms differentiating methanotrophic and methanogenic archaea. PLOS Biology, 20(1), e3001508. https://doi.org/10.1371/journal.pbio.3001508
- Metcalfe, K. S., Murali, R., Mullin, S. W., Connon, S. A., & Orphan, V. J. (2021). Experimentally-validated correlation analysis reveals new anaerobic methane oxidation partnerships with consortium-level heterogeneity in diazotrophy. The ISME Journal, 15(2), Article 2. https://doi.org/10.1038/s41396-020-00757-1
- Choi, H. M. T., Schwarzkopf, M., Fornace, M. E., Acharya, A., Artavanis, G., Stegmaier, J., Cunha, A., & Pierce, N. A. (2018). Third-generation in situ hybridization chain reaction: Multiplexed, quantitative, sensitive, versatile, robust. Development, 145(12), dev165753. https://doi.org/10.1242/dev.165753
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J.-Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P., & Cardona, A. (2012). Fiji: An open-source platform for biological-image analysis. Nature Methods, 9(7), Article 7. https://doi.org/10.1038/nmeth.2019
